
. DOI: 10.1172/JCI170369")
Credit: Journal of Clinical Investigations (2024). DOI: 10.1172/JCI170369
A study conducted by researchers at Baylor College of Medicine and collaborating institutions reveals the molecular events that lead to osteogenesis imperfecta type V, a form of bone fragility caused by a mutation in the IFITM5 gene.
The mutation blocks the normal development of bone stem cells into mature cells that would form healthy bones. Instead, the mutation leads to the formation of bones that are extremely brittle. Children with this disorder suffer recurrent bone fractures, bone deformities, chronic pain and other complications. The results, published in The Journal of Clinical Investigationsoffer new opportunities for the development of therapies for this currently untreatable disease.
“Bone fragility disorders, also called osteogenesis imperfecta (OI), are a group of rare disorders that affect connective tissues – tissues like bone that support and protect other tissues in the body,” said Brendan Lee, PhD, professor, chair and Robert and Janice McNair Endowed Chair in Molecular and Human Genetics at Baylor. He is also a member of Baylor’s Dan L Duncan Comprehensive Cancer Center and part of Texas Children’s Hospital. “Most types of OI are caused by gene mutations that disrupt collagen synthesis or processing, but not OI type V.”
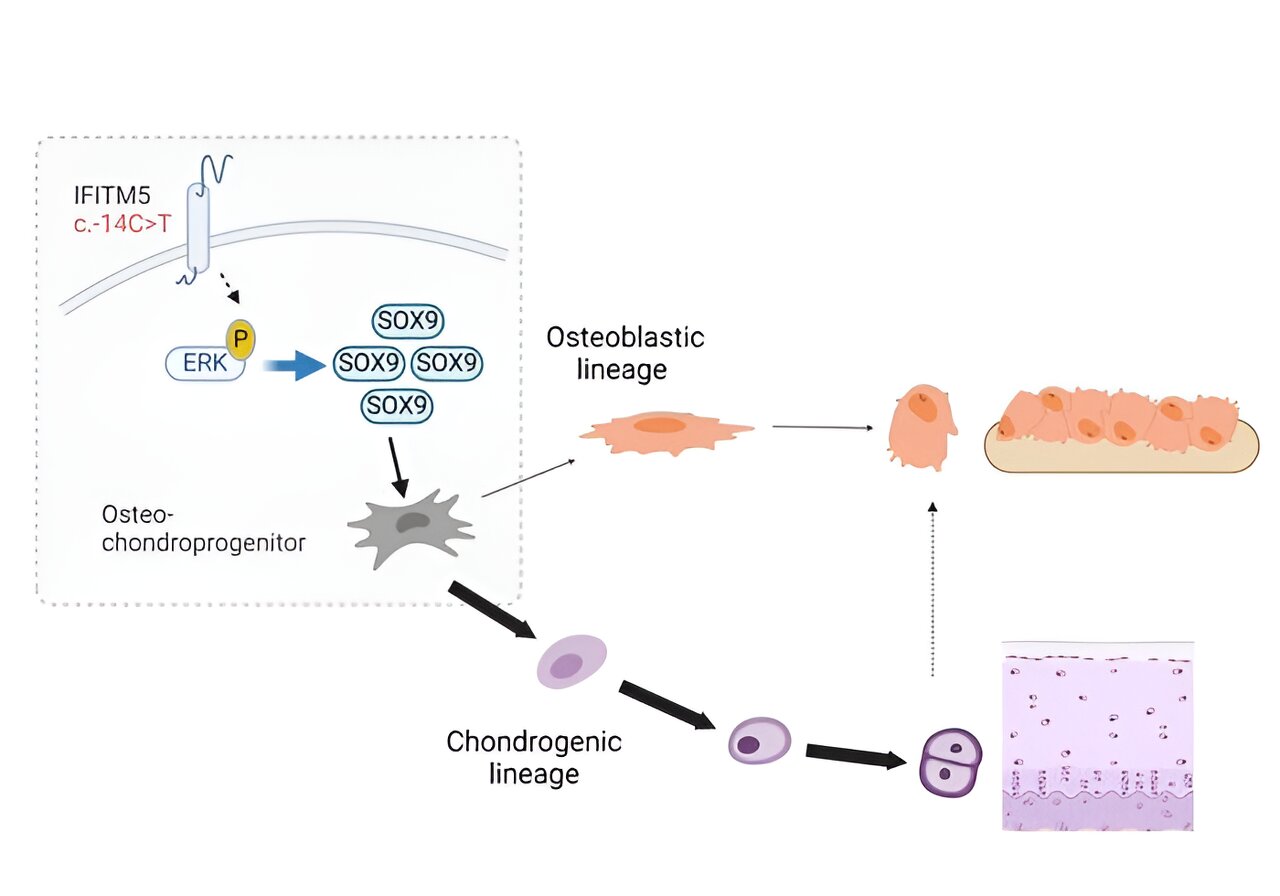
“OI type V is unique because all patients have exactly the same mutation in the IFITM5 gene that is associated with the disease,” said the paper’s lead author, Dr. Ronit Marom, assistant professor of molecular and human genetics at Baylor and Texas Children’s. “This mutation leads to the production of a longer IFITM5 protein; however, the biological function of this protein in bone and why this mutation leads to OI were not well understood.”
The researchers developed an animal model for OI type V by genetically modifying mice to express the mutated gene at specific stages of bone development. The genetically modified mice displayed most of the features of the human disease, allowing analysis of the underlying molecular mechanisms.
The team discovered that the IFITM5 mutation acts at the level of bone stem cells, altering the normal process that leads to bone formation. “Bone stem cells are leaders in the formation of the skeleton during development and in bone healing after a fracture – first they form cartilage, which then turns into bone,” Lee said.
The IFITM5 mutation in mice disrupts this process. Instead of developing from cartilage to bone, precursor cells form oversized cartilage calluses where new bone should be.
“Our findings help explain what we see in patients with OI type V. Not only do they have bones that break easily, but when stem cells try to heal them, they form large cartilage calluses instead of bone,” Lee said. “It’s as if the stem cells don’t finish their job, they get caught in a loop to preferentially form cartilage instead of maturing into bone.”
“Until now, we thought OI was the result of abnormal bone development. It was exciting to discover that OI type V is actually the result of abnormal differentiation of a common stem cell, leading to an imbalance in both cartilage and bone development,” said Marom.
The team also identified two important molecular factors that cause this bone maturation defect.
“The ERK/MAP kinase signaling pathway and the transcription factor SOX9 were both significantly increased,” said Marom. “Interestingly, when we inactivated either ERK/MAP kinase signaling or SOX9 using pharmacological or genetic approaches, we were able to restore normal bone development in mutant models. These findings not only shed light on the mechanism of OI type V, they will also facilitate the development of future therapies for this disease.”
“In addition, the fact that all patients with OI type V have the same IFITM5 mutation could prove to be an advantage for gene therapies targeting the mutated gene,” Lee said. “Gene therapy to treat the IFITM5 mutation would work in all patients.”
This study is another example of the value of rare disease studies for better understanding and treatment of common diseases.
“Understanding how OI type V develops provides new insights into similar but more common skeletal diseases such as osteoporosis and could also lead to improved treatments,” Lee said.
More information:
Ronit Marom et al., The IFITM5 mutation in osteogenesis imperfecta type V is associated with an ERK/SOX9-dependent osteoprogenitor differentiation defect, Journal of Clinical Investigations (2024). DOI: 10.1172/JCI170369
Provided by Baylor College of Medicine
Quote: Bone stem cells with IFITM5 mutation get caught in a loop that leads to osteogenesis imperfecta type V (June 26, 2024), accessed June 26, 2024 from https://medicalxpress.com/news/2024-06-bone-stem-cells-ifitm5-mutation.html
This document is subject to copyright. Except for the purposes of private study or research, no part of it may be reproduced without written permission. The contents are for information purposes only.



